The AAV Gene Therapy Regulatory Landscape
There are currently 359 candidates that are using Adeno-Associated Virus (AAV) technology to deliver genes to tissues for 133 diseases. With very few having received approval by a regulatory body, competition for late-stage candidates is particularly high. Below is a summary of recent approvals and their implications for the AAV market.
FDA
Currently, the FDA has approved two AAV gene therapies: Spark Therapeutics’ Luxturna for retinal dystrophy in 2017 and Novartis’ Zolgensma for spinal muscular atrophy in 2019. There have been no recent AAV gene therapy approvals by the FDA, however, last week, CSL Behring and uniQure’s etranacogene dezaparvovec was given Priority Review by the FDA for the treatment of Haemophilia B using AAV5. Priority Review status expedites the review process with the aim of completing the review within 6 months in comparison to the usual 10 months to allow for significant unmet treatment need to be met. This suggests that we will expect a regulatory decision on etranacogene dezaparvovec for the treatment of Haemophilia B by 2022 year end.
In March this year, the FDA lifted a clinical hold on a Phase III trial of Pfizer and Sangamo’s giroctocogene fitelparvovec for the treatment of Haemophilia A using rAAV6. The trial was put on hold in November 2021 due to safety concerns surrounding blood clots; Pfizer and Sangamo have announced that they will continue the pause to adjust the trial protocol but will resume in Q3 2022, with results expected in H2 2023.
In January this year however, BioMarin released positive Phase III data on its own AAV5 delivered gene therapy valoctocogene roxaparvovec (Roctavian) for the treatment of Haemophilia A, a competitor to Pfizer and Sangamo’s candidate; it is expected that the BioMarin will submit this data to the FDA as a resubmission in Q2 2022 after the FDA initially rejected the approval application in 2020, requesting further data on two-year annualised bleeding rate.
EMA
BioMarin’s valoctocogene roxaparvovec has also been under review by the EMA since Q3 2021 after BioMarin resubmitted the application and a regulatory decision by the EMA is expected in H1 2022.
The EMA regulatory landscape has seen more recent updates than the FDA space as last week PTC Therapeutics’ PTC-AADC (Upstaza) for the treatment of aromatic L-amino acid decarboxylase (AADC) deficiency was given a positive opinion by the Committee for Medicinal Products for Human Use (CHMP) and therefore recommended for approval by the EMA.
This AAV therapy uses an AAV2 vector to deliver the correct functioning form of dopa decarboxylase (DDC) into the brain; the treatment uses stereotactic neurosurgery and MRI to inject and deliver the therapy directly into the putamen. This will be the third AAV gene therapy approval by the EMA, following Luxturna in 2018 and Zolgensma in 2020. As the treatment requires a specialist cannula called SmartFlow® for administration, medical device approval was also needed prior to drug marketing authorisation.
The CE mark required by EU countries for medical devices was obtained in 2018 and 501(k) clearance by the FDA in 2011, however due to the additional regulation needed for the medical device, this could potentially delay any regulatory processes; PTC Therapeutics aims to submit a Biologics Licence Application (BLA) for FDA approval in the second quarter of 2022.
As well as being under FDA Priority Review, CSL Behring and uniQure’s etranacogene dezaparvovec is currently under review by the EMA’s Accelerated Assessment protocol which allows the review to take place in 150 days, rather than the standard 210 days. Accelerated Assessment was granted to etranacogene dezaparvovec by the CHMP in December 2021, with the Marketing Authorisation Application submitted at the end of March 2022; a decision by the EMA on the Haemophilia B therapy can be expected in Q3 2022.
Conclusions
Since the approvals of Luxturna and Zolgensma by the FDA and EMA in the late 2010s, there has been a pause in AAV gene therapy authorisations by the regulatory bodies. This trend appears to be changing however, with a number of AAV candidates having either submitted to the FDA and EMA for approval, or expecting results from pivotal late phase clinical trials that could lead to a submission.
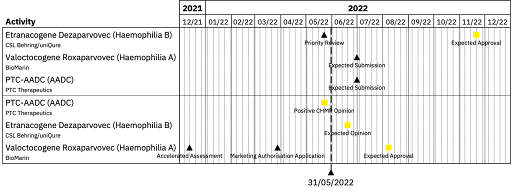
The Airfinity AAV gene therapy report monitors the clinical progression of AAV candidates, highlighting when candidates move through phases, as well as providing analysis on regulatory applications, designations and changes, and providing deep dive analysis on a range of topics including AAV serotype use and market growth.
Get in touch to discuss access to our AAV gene therapy report.